
Všetok obsah iLive je lekársky kontrolovaný alebo kontrolovaný, aby sa zabezpečila čo najväčšia presnosť faktov.
Máme prísne smernice týkajúce sa získavania zdrojov a len odkaz na seriózne mediálne stránky, akademické výskumné inštitúcie a vždy, keď je to možné, na lekársky partnerské štúdie. Všimnite si, že čísla v zátvorkách ([1], [2] atď.) Sú odkazmi na kliknutia na tieto štúdie.
Ak máte pocit, že niektorý z našich obsahov je nepresný, neaktuálny alebo inak sporný, vyberte ho a stlačte kláves Ctrl + Enter.
Treacher Collinsov syndróm
Lekársky expert článku

Posledná kontrola: 04.07.2025

Vnútromaternicové poruchy v procesoch vývoja kostí spôsobujú závažné kraniofaciálne deformity a jednou z odrôd takejto patológie je syndróm Treacher-Collins (TCS) alebo mandibulofasciálna, teda maxilofaciálna dysostóza.
Kód choroby podľa ICD 10: trieda XVII (vrodené anomálie, deformácie a chromozomálne poruchy), Q75.4 - mandibulofaciálna dysostóza.
Príčiny Treacher Collinsov syndróm
Tento syndróm bol pomenovaný po vynikajúcom britskom oftalmológovi Edwardovi Treacherovi Collinsovi, ktorý pred viac ako sto rokmi opísal hlavné črty tejto patológie. Európski lekári však tento typ anomálie tvárových a čeľustných kostí častejšie nazývajú Franceschettiho chorobou alebo syndrómom – na základe rozsiahleho výskumu švajčiarskeho oftalmológa Adolfa Franceschettiho, ktorý v polovici minulého storočia zaviedol termín „mandibulofasciálna dysostóza“. V lekárskych kruhoch sa používa aj názov Franceschettiho-Collinsov syndróm.
Treacher Collinsov syndróm je spôsobený mutáciami v géne TCOF1 (na chromozómovom lokuse 5q31.3-33.3), ktorý kóduje nukleolárny fosfoproteín zodpovedný za tvorbu kraniofaciálnej časti ľudského embrya. V dôsledku predčasného poklesu množstva tohto proteínu dochádza k narušeniu biogenézy a funkcií rRNA. Podľa genetikov z výskumného programu Human Genome tieto procesy vedú k zníženiu proliferácie embryonálnych buniek neurálnej lišty - hrebeňa pozdĺž neurálnej drážky, ktorá sa počas embryonálneho vývoja uzatvára do neurálnej trubice.
Tvorba tkanív tváre nastáva v dôsledku transformácie a diferenciácie buniek hornej (hlavovej) časti neurálnej lišty, ktoré migrujú pozdĺž neurálnej trubice do oblasti prvého a druhého branchiálneho oblúka embrya. A nedostatok týchto buniek spôsobuje kraniofaciálne deformácie. Kritické obdobie pre výskyt anomálií je od 18. do 28. dňa po oplodnení. Po ukončení migrácie buniek neurálnej lišty (v štvrtom týždni tehotenstva) sa vytvoria takmer všetky voľné mezenchymálne tkanivá v oblasti tváre, ktoré sa neskôr (od 5. do 8. týždňa) diferencujú na kostrové a spojivové tkanivá všetkých častí tváre, krku, hrtana, ucha (vrátane vnútorného ucha) a budúcich zubov.
Patogenézy
Patogenéza syndrómu Treacher-Collins je často familiárna a anomália sa dedí autozomálne dominantným spôsobom, hoci existujú prípady autozomálne recesívneho prenosu defektu (s mutáciami v iných génoch, najmä POLR1C a POLR1D). Najnepredvídateľnejšou vecou na maxilofaciálnej dysostóze je, že mutáciu dedia deti iba v 40 – 48 % prípadov. To znamená, že u 52 – 60 % pacientov nie sú príčiny syndrómu Treacher-Collins spojené s prítomnosťou anomálie v rodine a predpokladá sa, že patológia sa vyskytuje v dôsledku sporadických génových mutácií de novo. S najväčšou pravdepodobnosťou sú nové mutácie dôsledkom teratogénnych účinkov na plod počas tehotenstva.
Medzi teratogénne príčiny tohto syndrómu odborníci uvádzajú veľké dávky etanolu (etylalkoholu), žiarenie, cigaretový dym, cytomegavírus a toxoplazmu, ako aj herbicídy na báze glyfosátu (Roundal, Glyfor, Tornado atď.). Zoznam iatrogénnych faktorov zahŕňa lieky proti akné a seboree s kyselinou 13-cis-retínovou (Izotretinoín, Accutane); antikonvulzívum Fenytoín (Dilantin, Epanutín); psychotropné lieky Diazepam, Valium, Relanium, Seduxen.
Príznaky Treacher Collinsov syndróm
Klinické príznaky mandibulofasciálnej dysostózy a stupeň ich prejavu závisia vo väčšine prípadov od charakteristík prejavu génových mutácií. A prvé príznaky tejto anomálie sú vo väčšine prípadov viditeľné u dieťaťa bezprostredne po narodení: tvár so syndrómom Treacher-Collins má charakteristický vzhľad. Morfologické anomálie sú navyše zvyčajne bilaterálne a symetrické.
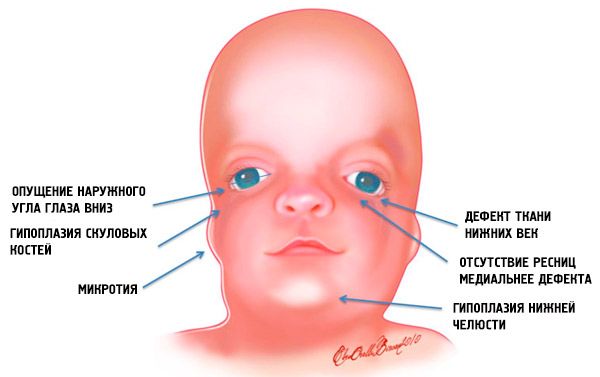
Najzreteľnejšie príznaky syndrómu Treacher-Collins sú:
- nedostatočný vývoj (hypoplázia) tvárových kostí lebky: zygomatické, zygomatické výbežky čelovej kosti, laterálne pterygoidné platničky, paranazálne dutiny, dolná čeľusť a výbežky kostných epifýz (kondyly);
- nedostatočný vývoj kostí dolnej čeľuste (mikrognatia) a tupejší uhol dolnej čeľuste ako zvyčajne;
- nos má normálnu veľkosť, ale javí sa ako veľký kvôli hypoplázii nadočnicových oblúkov a nedostatočnému vývoju alebo absencii zygomatických oblúkov v temporálnej oblasti;
- očné štrbiny smerujú nadol, to znamená, že tvar očí je abnormálny, pričom vonkajšie kútiky klesajú nadol;
- defekty dolných viečok (kolobóm) a čiastočná absencia mihalníc na nich;
- nepravidelne tvarované ušnice so širokou škálou odchýlok vrátane ich umiestnenia v rohu dolnej čeľuste, absencie lalokov, slepých fistúl medzi tragusom ucha a kútikom úst atď.;
- zúženie alebo uzavretie (atrézia) vonkajšieho zvukovodu a anomálie stredoušných kostičiek;
- absencia alebo hypoplázia príušných slinných žliaz;
- hypoplázia hltana (zúženie hltana a dýchacích ciest);
- nezrastenie tvrdého podnebia (rázštep podnebia), ako aj absencia, skrátenie alebo nehybnosť mäkkého podnebia.
Takéto anatomické anomálie majú vo všetkých prípadoch komplikácie. Ide o funkčné poruchy sluchu vo forme prevodovej straty sluchu alebo úplnej hluchoty; zrakové postihnutie v dôsledku nesprávneho vývoja očných buliev; defekty podnebia spôsobujú ťažkosti s kŕmením a prehĺtaním. Existujú poruchy zubného zhryzu (maloklúzia) spojené s defektmi čeľuste, čo zase spôsobuje problémy so žuvaním a artikuláciou. Patológie mäkkého podnebia vysvetľujú nosový hlas.
Komplikácie a následky
Dôsledky maxilofaciálnych anomálií pri syndróme Treacher Collins sú také, že pri narodení sú intelektuálne schopnosti dieťaťa normálne, ale v dôsledku sluchových porúch a iných porúch sa pozoruje sekundárna mentálna retardácia.
Okrem toho deti s takýmito chybami akútne pociťujú svoju menejcennosť a trpia, čo negatívne ovplyvňuje ich nervový systém a psychiku.
Diagnostika Treacher Collinsov syndróm
Postnatálna diagnostika Treacher-Collinsovho syndrómu je v podstate založená na klinických príznakoch. Kraniofaciálna dysostóza sa dá ľahko identifikovať, keď je syndróm plne expresívny, ale ak sú prítomné minimálne vyjadrené príznaky patológie, môžu vzniknúť problémy so stanovením správnej diagnózy.
V tomto prípade by sa mala venovať osobitná pozornosť posúdeniu všetkých funkcií spojených s anomáliami, najmä tých, ktoré ovplyvňujú dýchanie (kvôli riziku spánkového apnoe). Mala by sa tiež posúdiť a monitorovať účinnosť kŕmenia a saturácia hemoglobínu kyslíkom.
Neskôr, na 5. – 6. deň po narodení, bude potrebné určiť rozsah poškodenia sluchu pomocou audiologického vyšetrenia, ktoré by sa malo vykonať v pôrodnici.
Predpísané je vyšetrenie, počas ktorého sa vykonáva inštrumentálna diagnostika fluoroskopiou kraniofaciálnej dysmorfológie; pantomografia (panoramatický röntgen kostných štruktúr tvárovej lebky); kompletná kraniálna počítačová tomografia v rôznych projekciách; CT alebo MRI mozgu na určenie stavu vnútorného zvukovodu.
Najskoršia – prenatálna – diagnostika maxilofaciálnych anomálií pri prítomnosti Treacher Collinsovho syndrómu v rodinnej anamnéze je možná biopsiou choriových klkov v 10. – 11. týždni tehotenstva (zákrok ohrozuje potrat a infekciu maternice).
Krvné testy sa odoberajú aj členom rodiny; v 16. – 17. týždni tehotenstva sa analyzuje plodová voda (transabdominálna amniocentéza); v 18. – 20. týždni tehotenstva sa vykonáva fetoskopia a odoberá sa krv z fetálnych ciev placenty.
Najčastejšie sa však ultrazvuk používa pri prenatálnej diagnostike tohto syndrómu u plodu (v 20. – 24. týždni tehotenstva).
Aké testy sú potrebné?
Odlišná diagnóza
Tieto isté metódy používajú špecialisti, keď je potrebná diferenciálna diagnostika na rozpoznanie mierneho Treacher-Collinsovho syndrómu a jeho odlíšenie od iných vrodených anomálií kraniofaciálnych kostí, najmä: Apertovho, Crouzonovho, Nagerovho, Peters-Hewelsovho, Hellermann-Stephovho syndrómu, ako aj hemifaciálnej mikrozómie (Goldenharov syndróm), hypertelorizmu, predčasného zrastu lebečných stehov (kraniosynostóza) alebo zhoršeného zrastu tvárových kostí (kraniosynostóza).
Liečba Treacher Collinsov syndróm
Rovnako ako vo všetkých prípadoch geneticky podmienených vrodených chýb, liečba závažných foriem syndrómu Treacher-Collins je výlučne paliatívna, pretože na takéto patológie jednoducho neexistujú žiadne terapeutické metódy. Spektrum a stupeň deformácií pri tomto syndróme sú rozsiahle, a preto má povaha a intenzita lekárskeho zásahu tiež veľa možností.
Na korekciu a zlepšenie sluchu sa používajú načúvacie prístroje a na zlepšenie reči sa používajú logopedické sedenia.
Chirurgické zákroky sú potrebné už v ranom veku v závažných prípadoch zúženia dýchacích ciest (vykonáva sa tracheostómia) a hrtana (vykonáva sa gastrostómia na kŕmenie). Môže byť potrebná aj chirurgická korekcia podnebia.
Operácie predĺženia mandibuly sa vykonávajú vo veku 2-3 rokov alebo neskôr. Rekonštrukcia mäkkých tkanív zahŕňa korekciu kolobómu dolného viečka a plastickú chirurgiu ušných zubov.
Predpoveď
Aká je prognóza tejto patológie? Závisí od stupňa deformácie a intenzity symptómov. Treacher-Collinsov syndróm je celoživotná diagnóza.
 [ 25 ]
[ 25 ]