
Všetok obsah iLive je lekársky kontrolovaný alebo kontrolovaný, aby sa zabezpečila čo najväčšia presnosť faktov.
Máme prísne smernice týkajúce sa získavania zdrojov a len odkaz na seriózne mediálne stránky, akademické výskumné inštitúcie a vždy, keď je to možné, na lekársky partnerské štúdie. Všimnite si, že čísla v zátvorkách ([1], [2] atď.) Sú odkazmi na kliknutia na tieto štúdie.
Ak máte pocit, že niektorý z našich obsahov je nepresný, neaktuálny alebo inak sporný, vyberte ho a stlačte kláves Ctrl + Enter.
Alkaptonúria je vrodená enzýmová abnormalita
Lekársky expert článku

Posledná kontrola: 04.07.2025
Jedna z veľmi zriedkavých metabolických porúch, alkaptonúria, sa vzťahuje na vrodené anomálie v metabolizme aminokyseliny tyrozínu.
Tento syndróm sa môže nazývať aj deficit homogentizátoxidázy, homogentizinúria, dedičná ochronóza alebo ochorenie čierneho moču.[ 1 ]
Epidemiológia
Podľa štatistík nie je viac ako deväť prípadov alkaptonúrie na 1 milión ľudí. A vo väčšine európskych krajín je jeden prípad na 100 – 250 tisíc živonarodených detí.
Spomedzi európskych krajín je výnimkou Slovensko (najmä relatívne malý severozápadný región), kde je prevalencia alkaptonúrie jeden prípad na 19 000 novorodencov. Je to s najväčšou pravdepodobnosťou spôsobené tým, že medzi slovenskými rómskymi rodinami, ktoré tam žijú, je úroveň inbreedingu (manželstiev medzi bratrancami a sesternicami) najvyššia v Európe: 10 – 14 %. [ 2 ]
Príčiny alkaptonúria
Presné príčiny alkaptonúrie ako vrodenej poruchy katabolizmu (metabolického rozkladu) aromatickej (homocyklickej) α-aminokyseliny tyrozínu boli stanovené: tento typ metabolickej poruchy je dôsledkom homozygotných alebo zložených heterozygotných mutácií jedného z tisícov génov na chromozóme 3, presnejšie génu HGD v lokuse 3q21-q23 na dlhom ramene chromozómu. Tento gén kóduje nukleotidové sekvencie pečeňového enzýmu homogentisát-1,2-dioxygenáza [ 3 ] (tiež nazývaná oxidáza kyseliny homogentisovej alebo homogentisátoxidáza) - metaloproteínu obsahujúceho železo, ktorý je nevyhnutný pre jednu zo stupňov rozkladu tyrozínu v tele. [ 4 ], [ 5 ]
Alkaptonúria je teda poruchou enzýmu homogentisát-1,2-dioxygenáza, alebo presnejšie povedané, výsledkom jeho geneticky podmieneného nedostatku alebo úplnej absencie. [ 6 ]
Keďže ide o vrodený nedostatok enzýmu, alkaptonúria sa dedí autozomálne recesívne, to znamená, že na to, aby sa alkaptonúria u detí vyskytla, musia mať obaja rodičia modifikovaný gén pre tento enzým, pretože každý z nich odovzdá dieťaťu iba jednu kópiu génu z dvoch dostupných.
Podľa najnovších údajov existuje viac ako dvesto variantov modifikácie génu HGD a najčastejšie sa pozorujú missense mutácie, translokácia a splicing.
Rizikové faktory
Jediným rizikovým faktorom vzniku tejto vrodenej enzymopatie je jej prítomnosť v rodinnej anamnéze a dedičnosť dvoch modifikovaných kópií génu HGD, ak rodičia nevykazujú alkaptonúriu (riziko prenosu anomálie je 25 %), alebo jeden z rodičov má túto poruchu. [ 7 ]
Patogenézy
Tyrozín hrá kľúčovú úlohu v syntéze bielkovín, produkcii chromoproteínov – kožného pigmentu melanínu, ako aj hormónov štítnej žľazy a katecholamínových neurotransmiterov.
Mechanizmus regulácie množstva tyrozínu v bunkách je veľmi zložitý a telo normalizuje jeho nadmerný obsah jeho odbúravaním. Proces katabolizmu tyrozínu, rovnako ako všetky aromatické aminokyseliny, je viacstupňový a prebieha v niekoľkých fázach. Každá fáza metabolického odbúravania tyrozínu prebieha za účasti špecifického enzýmu a za vzniku medziproduktu.
Takže najprv sa aminokyselina rozloží na para-hydroxyfenylpyruvát, ktorý sa premení na alkaptón – 2,5-dihydroxyfenyloctovú alebo kyselinu homogentisovú. Potom by sa alkaptón mal premeniť na kyselinu maleoctovú, ale to sa nestane. [ 8 ]
A patogenéza alkaptonúrie spočíva v zastavení biochemických reakcií katabolizmu tyrozínu v štádiu tvorby kyseliny homogentisovej: na jej rozklad jednoducho nie je potrebný žiadny enzým – homogentisátoxidáza.
Kyselina homogentisová nie je v tele využitá a môže sa hromadiť vylučovaním obličkami. Okrem toho sa oxiduje na benzochinoacetát (kyselina benzochinónoctová), ktorý väzbou na molekuly tkanív a telesných tekutín vytvára biopolymérne zlúčeniny sfarbené ako melanín.
Hromadenie týchto medziproduktov v tkanive vedie k narušeniu kolagénovej štruktúry chrupavkového tkaniva, čo znižuje jeho elasticitu – s výskytom mnohých klinických príznakov alkaptonúrie a rozvojom komplikácií.
Príznaky alkaptonúria
Alkaptonúria u novorodencov a dojčiat sa vyznačuje stmavnutím moču. Pri kontakte so vzduchom sa moč na plienkach, zavinovačke a spodnej bielizni sfarbí do tmavohnedého odtieňa; je to spôsobené hromadením a uvoľňovaním kyseliny homogentisovej, ktorá sa oxiduje na benzochinoacetát. [ 9 ]
Pri absencii iných príznakov sa alkaptonúria u malých detí často nerozpozná včas, pretože moč môže po niekoľkých hodinách močenia stmavnúť. Podľa niektorých údajov je v klinickom prostredí identifikovaná iba pätina detí mladších ako 12 mesiacov, ktoré sa narodili s nedostatkom tohto enzýmu. Preto je veľmi dôležité, aby rodičia venovali pozornosť starostlivosti o svoje dojčatá.
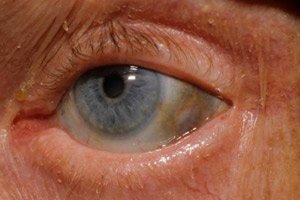
Medzi skoré príznaky patrí aj pigmentácia (modrosivá farba) bielka očí a chrupaviek uší a nosa, čo sa často nazýva ochronóza.[ 10 ]
Postupom času sa objavujú ďalšie príznaky:
- silná pigmentácia kože na lícnych kostiach, v podpazuší a na genitáliách;
- zafarbenie oblečenia pri kontakte s potenými oblasťami tela;
- záchvaty všeobecnej slabosti;
- chrapľavý hlas.
Treba mať na pamäti, že alkaptonúria a ochronóza, ako je uvedené vyššie, sú synonymné názvy pre tú istú poruchu katabolizmu tyrozínu.
Javorový sirupový močový syndróm a alkaptonúria. Vrodený javorový sirupový močový syndróm alebo leucinóza je tiež metabolická porucha, má rovnaký vzorec dedičnosti a dokonca sa mutácie vyskytujú na rovnakom chromozóme, ale ovplyvňujú gén kódujúci enzýmový komplex rozvetvenej α-ketokyseliny dehydrogenázy. Z tohto dôvodu telo nedokáže rozložiť určité zložky bielkovín, najmä aminokyseliny leucín, izoleucín a valín. Pri tomto ochorení má moč (a ušný maz) sladkú vôňu; okrem toho klinický obraz tohto typu organickej acidémie zahŕňa hypopigmentáciu, kolísanie krvného tlaku, záchvaty, vracanie a hnačku, pokles hladiny glukózy v krvi, ketoacidózu, halucinácie atď. Úmrtnosť u detí je pomerne vysoká; u dospelých môže bez liečby dôjsť ku kóme a smrti v dôsledku mozgového edému.
Albinizmus a alkaptonúriu „spája“ iba tyrozín. Albinizmus, vrátane okulokutánneho, je spôsobený genetickými mutáciami, ktoré ovplyvňujú produkciu pigmentu melanínu. Vrodené zmeny sa zaznamenávajú v géne TYR na chromozóme 11 (11q14.3), ktorý kóduje tyrozinázu, melanozómový enzým obsahujúci meď, ktorý je potrebný na tvorbu kožného pigmentu na základe produktov metabolizmu tyrozínu. Toto ochorenie je oveľa častejšie ako alkaptonúria.
Komplikácie a následky
Dôsledky a komplikácie alkaptonúrie, spôsobené pôsobením medziproduktov tyrozínu – kyseliny homogentisovej a benzochinónoctovej – sa objavujú v dôsledku ukladania reaktívnych pigmentovaných polymérov, deštrukcie kolagénových fibríl a zhoršenia stavu chrupavky (so znížením jej odolnosti voči mechanickému namáhaniu).
V priebehu rokov, v dospelosti, sa vyvíja degeneratívna artritída a osteoartróza veľkých kĺbov (bedrových, sakroiliakálnych a kolenných); medzistavcové priestory sa zužujú (najmä v bedrovej a hrudnej chrbtici) – s kalcifikáciou a tvorbou osteofytov; hustota tkaniva subchondrálnych kostných platničiek sa znižuje a podkladové kosti môžu podliehať patologickej prestavbe s tvorbou výrastkov a deformácií. [ 11 ]
V dôsledku rovnakej kalcifikácie sa môže pozorovať poškodenie srdcových chlopní (aortálnej a mitrálnej) a koronárnych artérií – s príznakmi ischemickej choroby srdca, ako aj tvorba kameňov v obličkách a prostate. [ 12 ], [ 13 ]
Diagnostika alkaptonúria
Diagnóza vrodených metabolických porúch je zvyčajne založená na štúdiu biologických tekutín tela.
Na základe akých testov a reakcií možno diagnostikovať alkaptonúriu? Na zistenie kyseliny homogentisovej a stanovenie jej hladiny (normálna – 20 – 30 mg za deň, zvýšená – 3 – 8 g) sú potrebné testy moču. Vzorka moču sa vyšetruje plynovou chromatografiou alebo hmotnostnou spektrometriou s použitím kvapalinovej chromatografie; možný je aj skríningový test na prítomnosť chloridu železa v moči. [ 14 ]
Existuje aj metóda rýchlej diagnostiky – stanovenie alkaptónu v zaschnutých škvrnách moču na papieri (podľa intenzity farby).
Pri objasňovaní diagnózy zahŕňa inštrumentálna diagnostika (rádiografia) identifikáciu rádiologických príznakov osteoartrózy a iných kĺbových patológií u pacientov.
Diagnóza sa potvrdzuje molekulárno-genetickými metódami diagnostikovania dedičných chorôb, ako sú genetické testovanie a sekvenovanie DNA. [ 15 ]
Odlišná diagnóza
Diferenciálna diagnostika zahŕňa hemochromatózu a akútne zlyhanie pečene novorodencov, melaninúriu, akútnu intermitentnú porfýriu, hemofagocytárnu lymfohistiocytózu, primárnu mitochondriálnu patológiu, reumatoidnú artritídu, ankylozujúcu spondylitídu.
Komu sa chcete obrátiť?
Liečba alkaptonúria
Hlavnou liečbou alkaptonúrie je perorálne podávanie veľkých dávok (najmenej 1000 mg denne) kyseliny askorbovej. U detí to zvyšuje vylučovanie kyseliny homogentisovej močom a u dospelých to znižuje obsah jej derivátu, kyseliny benzochinónoctovej, v moči a spomaľuje jej väzbu na štruktúry spojivového tkaniva kĺbov a kolagénu. [ 16 ]
Západoeurópske kliniky testujú liek Nitizinón (Orfalin), liek zo skupiny metabolitov, ktorý inhibuje druhú fázu katabolizmu tyrozínu: transformáciu para-hydroxyfenylpyruvátu na kyselinu homogentisovú. Použitie tohto farmakologického činidla však vedie k akumulácii tyrozínu a môže spôsobiť závažné vedľajšie účinky vrátane zákalu rohovky a fotofóbie, krvácania z nosa a žalúdka, zlyhania pečene, zmien v krvi atď. Napriek tomu je v Spojených štátoch Nitizinón schválený Úradom pre kontrolu potravín a liečiv (FDA) na liečbu tyrozinémie typu I. [17 ], [ 18 ]
Preto sa pri problémoch s kĺbmi spôsobených alkaptonúriou vykonáva fyzioterapia – cvičebná terapia na zvýšenie svalovej sily a zlepšenie pohyblivosti kĺbov, balneoterapia a peloidná terapia na obmedzenie bolesti.
Hoci tyrozín sa nielenže dodáva z potravy, ale sa aj produkuje v tele, pacientom s alkaptonúriou sa odporúča dodržiavať diétu s nízkym obsahom bielkovín a obmedziť konzumáciu potravín bohatých na tyrozín, predovšetkým hovädzieho a bravčového mäsa, mliečnych výrobkov (najmä syrov), strukovín, orechov a semien.
Prevencia
Prevencia génových mutácií je nemožná, ale na zabránenie narodenia detí s vysokým rizikom vrodených porúch existuje lekársko-genetické poradenstvo, ktoré je nevyhnutné pred plánovaným tehotenstvom pre páry, ktorých rodinná anamnéza zahŕňa dedičné choroby. [ 19 ]
Predpoveď
Smrteľné následky alkaptonúrie sú veľmi zriedkavé a smrť môže byť spôsobená závažnými komplikáciami postihujúcimi srdce a obličky. Celková dĺžka života ľudí s alkaptonúriou je teda dobrá.
Kvalita života sa však znižuje v dôsledku intenzívnej bolesti v kĺboch alebo chrbtici s výrazným obmedzením pohyblivosti, často progresívnej.