
Všetok obsah iLive je lekársky kontrolovaný alebo kontrolovaný, aby sa zabezpečila čo najväčšia presnosť faktov.
Máme prísne smernice týkajúce sa získavania zdrojov a len odkaz na seriózne mediálne stránky, akademické výskumné inštitúcie a vždy, keď je to možné, na lekársky partnerské štúdie. Všimnite si, že čísla v zátvorkách ([1], [2] atď.) Sú odkazmi na kliknutia na tieto štúdie.
Ak máte pocit, že niektorý z našich obsahov je nepresný, neaktuálny alebo inak sporný, vyberte ho a stlačte kláves Ctrl + Enter.
Lissencefália mozgu
Lekársky expert článku

Posledná kontrola: 12.07.2025
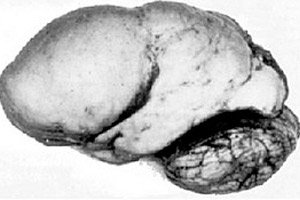
Medzi organickými mozgovými patológiami vyniká taká vrodená anomália vývoja mozgu, ako je lissencefália, ktorej podstata spočíva v takmer hladkom povrchu kôry jej hemisfér - s nedostatočným počtom závitov a ryh. [ 1 ]
Pri úplnej absencii konvolúcií sa definuje agýria a prítomnosť niekoľkých širokých plochých konvolúcií sa nazýva pachygyria. Tieto defekty, rovnako ako niektoré iné redukčné deformácie mozgu, majú v ICD-10 kód Q04.3.
Epidemiológia
Podľa štatistík o zriedkavých ochoreniach pripadá 1 – 1,2 prípadu lissencefálie na 100 tisíc novorodencov. [ 2 ], [ 3 ]
Podľa niektorých údajov sa u detí s Millerovým-Diekerovým syndrómom pozoruje až 25 – 30 % prípadov klasickej lissencefálie; bodové mutácie a delécie génov LIS1 a DCX sa detegujú u takmer 85 % pacientov. [ 4 ]
Genetické štúdie 17 génov spojených s lissencefáliou ukázali, že mutácia alebo delécia LIS1 je zodpovedná za 40 % pacientov a 23 % je spojených s mutáciou DCX, nasledujú TUBA1A (5 %) a DYNC1H1 (3 %).[ 5 ]
Príčiny lissencefália
Všetky známe príčiny vzniku mozgovej kôry (cortex cerebri) takmer alebo úplne bez závitov a ryh, ktoré zväčšujú „pracovnú plochu“ ľudského mozgu a zabezpečujú „produktivitu“ centrálneho nervového systému, sú spojené s poruchami jeho perinatálneho vývoja. To znamená, že u plodu sa vyvíja lissencefália. [ 6 ]
Neschopnosť tvoriť vrstvy mozgovej kôry plodu pri lissencefálii je výsledkom abnormálnej migrácie neurónov, ktoré ju tvoria, alebo predčasného ukončenia tohto procesu.
Tento proces, ktorý je nevyhnutný pre cerebrokortikálnu histogenézu, prebieha v niekoľkých štádiách od 7. do 18. týždňa tehotenstva. A vzhľadom na jeho zvýšenú citlivosť na genetické mutácie, ako aj na rôzne negatívne fyzikálne, chemické a biologické vplyvy, môže akákoľvek odchýlka od normy viesť k nesprávnej lokalizácii neurónov s možnou tvorbou zhrubnutej vrstvy sivej hmoty kôry bez charakteristickej štruktúry. [ 7 ]
V niektorých prípadoch je lissencefália u detí spojená s Millerovým-Diekerovým, Walkerovým-Warburgovým alebo Normanovým-Robertsovým syndrómom.
Prečítajte si tiež – Vývojové chyby mozgu
Rizikové faktory
Okrem mutácií v niektorých génoch patria medzi rizikové faktory narodenia dieťaťa s takouto závažnou chybou aj kyslíkové hladovanie (hypoxia) plodu; nedostatočné prekrvenie mozgu (hypoperfúzia); akútna mozgová príhoda vo forme perinatálnej mozgovej príhody; patológie placenty; vírusové infekcie tehotnej ženy (vrátane TORCH); [ 8 ] problémy s celkovým metabolizmom a funkciou štítnej žľazy; fajčenie, alkohol, psychotropné a omamné látky; užívanie viacerých liekov; zvýšené hladiny žiarenia. [ 9 ]
Patogenézy
Nie všetky prípady lissencefálie majú patogenézu spôsobenú chromozomálnymi abnormalitami a génovými mutáciami. Sú však známe niektoré gény, ktoré kódujú proteíny hrajúce dôležitú úlohu v správnom pohybe neuroblastov a neurónov pozdĺž radiálnych gliových buniek – za vzniku mozgovej kôry. A mutácie týchto génov vedú k tejto patológii. [ 10 ]
Ide najmä o sporadické mutácie (bez dedičnosti) génu LIS1 na chromozóme 17, ktorý reguluje cytoplazmatický motorický proteín mikrotubulov dyneín, ako aj gén DCX na chromozóme X, ktorý kóduje proteín doublecortin (lisencefalín-X). [ 11 ]. V prvom prípade odborníci definujú klasickú lissencefáliu (typ I), v druhom – viazanú na chromozóm X. [ 12 ]
Keď je gén FLN1, ktorý kóduje fosfoproteín filamín 1, odstránený, proces riadenej migrácie neurónov sa nemusí vôbec začať, čo vedie k úplnej absencii konvolúcií (agýria). [ 13 ]
V géne CDK5 boli identifikované mutácie, ktoré kódujú kinázový enzým – katalyzátor intracelulárneho metabolizmu, regulujúci bunkový cyklus v neurónoch CNS a zabezpečujúci ich normálnu migráciu počas prenatálneho formovania mozgových štruktúr.
Abnormálne zmeny v géne RELN na chromozóme 7, ktoré spôsobujú kortikálne gyrálne defekty pri Normanovom-Robertovom syndróme, vedú k nedostatku extracelulárneho glykoproteínu reelínu, ktorý je potrebný na reguláciu migrácie a umiestnenia nervových kmeňových buniek počas vývoja mozgovej kôry.[ 14 ], [ 15 ], [ 16 ]
Gén ARX kóduje nearistalensový homeoboxový proteín, transkripčný faktor, ktorý hrá dôležitú úlohu v prednom mozgu a iných tkanivách.[ 17 ] Deti s mutáciou ARX majú ďalšie príznaky, ako sú chýbajúce časti mozgu (agenéza corpus callosum), abnormálne genitálie a ťažká epilepsia.[ 18 ],[ 19 ]
S lissencefáliou bolo spojených niekoľko génov. Medzi tieto gény patria VLDLR, ACTB, ACTG1, TUBG1, KIF5C, KIF2A a CDK5.[ 20 ]
Cytomegalovírus (CMV) sa spája s rozvojom lissencefálie v dôsledku zníženého prekrvenia mozgu plodu. Závažnosť infekcie CMV závisí od gestačného veku. Včasná infekcia s väčšou pravdepodobnosťou spôsobí lissencefáliu, pretože k migrácii neurónov dochádza na začiatku tehotenstva.[ 21 ]
Okrem toho mechanizmus výskytu tejto anomálie zahŕňa neúplné alebo neskoršie zastavenie migrácie neurónov z periventrikulárnej generatívnej zóny do mozgovej kôry. A v takýchto prípadoch sa vyvíja buď neúplná lissencefália, alebo pachygyria, v ktorej sa tvorí niekoľko širokých drážok a záhybov (ale väčšina z nich chýba).
Príznaky lissencefália
Prvé príznaky tejto patológie (pri absencii predtým spomínaných syndrómov) sa nemusia objaviť ihneď po narodení, ale po jednom a pol až dvoch mesiacoch. A najčastejšie sa pozorujú nasledujúce klinické príznaky lissencefálie:
- svalová hypotónia, často kombinovaná so spastickou paralýzou;
- kŕče a generalizované tonicko-klonické záchvaty (vo forme opistotonusu);
- ťažká mentálna retardácia a rastová retardácia;
- zhoršenie neurologických a motorických funkcií.
Problémy s prehĺtaním spôsobujú ťažkosti s kŕmením dieťaťa. [ 22 ]
Vysoký stupeň neuromotorického poškodenia sa často prejavuje ako tetraplégia – paralýza všetkých končatín. Možná je deformácia rúk, prstov na rukách alebo nohách.
Pri Normanovom-Robertovom syndróme s lissencefáliou typu I sa pozorujú kraniofaciálne anomálie: ťažká mikrocefália, nízky sklon čela a vyčnievajúci široký koreň nosa, široko posadené oči (hyperterlorizmus), nedostatočný vývoj čeľustí (mikrognatia). [ 23 ]
Millerov-Diekerov syndróm môže byť tiež charakterizovaný abnormálne malou veľkosťou hlavy so širokým, vysokým čelom a krátkym nosom, priehlbinami v spánkoch (bitemporálne priehlbiny) a nízko nasadenými, deformovanými ušami.
Syndróm závažnej lissencefálie je charakterizovaný mikrocefaliou, zmenšením veľkosti očných buliev (mikroftalmia), v kombinácii s dyspláziou sietnice, obštrukčným hydrocefalom a chýbajúcim alebo hypoplastickým corpus callosum.
Komplikácie a následky
Medzi komplikácie tejto anomálie odborníci uvádzajú poruchu prehĺtania (dysfágiu) a gastroezofageálny reflux; refraktérnu (nekontrolovanú) epilepsiu; časté infekcie horných dýchacích ciest; pneumóniu (vrátane chronickej aspirácie).
Dojčatá s lissencefáliou môžu mať vrodené organické srdcové problémy vo forme defektu predsieňového septa alebo komplexnej srdcovej chyby s cyanózou (tetralógia Fallot). [ 24 ]
Dôsledky popôrodnej retardácie rastu vo väčšine prípadov vedú k úmrtiu do 24 mesiacov po narodení.
Diagnostika lissencefália
Diagnóza začína fyzickým vyšetrením dieťaťa, štúdiom anamnézy rodičov a anamnézy tehotenstva a pôrodu.
Počas tehotenstva môže byť potrebné vyšetrenie fetálnej DNA bez buniek, amniocentéza alebo odber choriových klkov. [ 25 ] Viac informácií nájdete v časti – Prenatálna diagnostika vrodených ochorení
Inštrumentálna diagnostika sa používa na vizualizáciu mozgových štruktúr a posúdenie ich funkcií:
- počítačová tomografia mozgu;
- magnetická rezonancia (MRI) mozgu;
- elektroencefalogram (EEG). [ 26 ]
Počas tehotenstva môže byť lissencefália na ultrazvuku plodu po 20-21 týždňoch podozrivá pri absencii parieto-okcipitálnych a kalkarinných drážok a anomálii Sylvianovej štrbiny mozgu.
Odlišná diagnóza
Vykonáva sa diferenciálna diagnostika s inými syndrómami vrodených mozgových chýb.
Existuje viac ako 20 typov lissencefálie, z ktorých väčšina spadá do 2 hlavných kategórií: klasická lissencefália (typ 1) a dlažobná lissencefália (typ 2). Každá kategória má podobné klinické prejavy, ale odlišné genetické mutácie.[ 27 ]
Vyšetrenie mozgu pri lissencefálii typu I ukazuje mozgovú kôru so štyrmi vrstvami namiesto šiestich, aké sa vyskytujú u zdravých pacientov, zatiaľ čo pri lissencefálii typu 2 je mozgová kôra dezorganizovaná a javí sa ako hrudkovitá alebo nodulárna v dôsledku úplného vytesnenia mozgovej kôry zhlukmi kortikálnych neurónov oddelených gliomezenchymálnym tkanivom. Pacienti mali tiež svalové a očné abnormality.
- Klasická lissencefália (typ 1):
- LIS1: Izolovaná lissencefália a Millerov-Diekerov syndróm (lissencefália spojená s tvárovou dysmorfizáciou). [ 28 ]
- LISX1: Mutácia génu DCX. V porovnaní s lissencefáliou spôsobenou mutáciami LIS1 vykazuje DCX šesťvrstvovú kôru namiesto štyroch.
- Izolovaná lissencefália bez iných známych genetických defektov
- Dlažobné kocky lissencefálie (typ 2):
- Walkerov-Warburgov syndróm
- Fukuyamov syndróm
- Ochorenie svalov, očí a mozgu
- Iné typy nemožno zaradiť do jednej z dvoch vyššie uvedených skupín:
- LIS2: Normanov-Robertsov syndróm, podobný lissencefálii typu I alebo Millerovmu-Diekerovmu syndrómu, ale bez delécie chromozómu 17.
- LIS3
- LISX2
Mikrolisencefália: Ide o kombináciu absencie normálneho kortikálneho zloženia a abnormálne malej hlavy. Deti s pravidelnou lissencefáliou majú pri narodení normálnu veľkosť hlavy. Deťom s malou veľkosťou hlavy pri narodení sa zvyčajne diagnostikuje mikrolisencefália.
Je tiež dôležité rozlišovať medzi lissencefáliou a polymikrogýriou, čo sú rôzne vývojové chyby mozgu.
Komu sa chcete obrátiť?
Liečba lissencefália
Lissencefália je nevyliečiteľná organická porucha, takže je možná iba podporná a symptomatická liečba. [ 29 ]
V prvom rade ide o použitie antikonvulzív a antiepileptík, ako aj o zavedenie gastrostomickej sondy do žalúdka (ak dieťa nedokáže samostatne prehĺtať). Masáž je užitočná.
V prípadoch závažného hydrocefalu sa odoberá mozgovomiechový mok.
Prevencia
Odborníci odporúčajú, aby budúci rodičia vyhľadali genetické poradenstvo a aby sa tehotné ženy včas zaregistrovali u pôrodníkov a gynekológov a absolvovali všetky plánované vyšetrenia.
Predpoveď
U detí s lissencefáliou závisí prognóza od jej stupňa, ale najčastejšie mentálny vývoj dieťaťa nepresahuje úroveň štyroch až piatich mesiacov. A všetky deti s touto diagnózou trpia ťažkými psychomotorickými poruchami a ťažko liečiteľnou epilepsiou. [ 30 ]
Podľa NINDS (Americký národný inštitút neurologických porúch a mozgovej príhody) je maximálna dĺžka života ľudí s lissencefáliou približne 10 rokov.