
Všetok obsah iLive je lekársky kontrolovaný alebo kontrolovaný, aby sa zabezpečila čo najväčšia presnosť faktov.
Máme prísne smernice týkajúce sa získavania zdrojov a len odkaz na seriózne mediálne stránky, akademické výskumné inštitúcie a vždy, keď je to možné, na lekársky partnerské štúdie. Všimnite si, že čísla v zátvorkách ([1], [2] atď.) Sú odkazmi na kliknutia na tieto štúdie.
Ak máte pocit, že niektorý z našich obsahov je nepresný, neaktuálny alebo inak sporný, vyberte ho a stlačte kláves Ctrl + Enter.
Choroba Charcot-Marie-Tooth.
Lekársky expert článku

Posledná kontrola: 12.07.2025
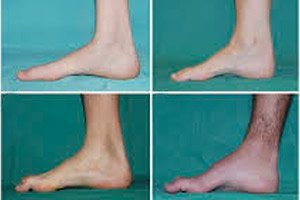
Peroneálna svalová atrofia, Charcot-Marie-Toothov syndróm alebo choroba, je skupina chronických dedičných ochorení s poškodením periférnych nervov.
Podľa ICD-10 je v časti o chorobách nervového systému kódom pre toto ochorenie G60.0 (hereditárna motorická a senzorická neuropatia). Je tiež zaradená do zoznamu orphan chorôb.
Epidemiológia
Podľa klinických štatistík je prevalencia všetkých typov Charcot-Marie-Toothovej choroby na 100 tisíc obyvateľov 19 prípadov (podľa iných zdrojov jeden prípad na 2,5 – 10 tisíc obyvateľov).
CMT typu 1 predstavuje približne dve tretiny prípadov (jeden prípad na 5 000 až 7 000 obyvateľov) a takmer 70 % z nich je spojených s duplikáciou génu PMP22. Týmto typom ochorenia trpí viac ako 1,2 milióna ľudí na celom svete.
Výskyt CMT typu 4 sa odhaduje na 1 – 5 prípadov na 10 000 detí. [ 1 ]
Príčiny Choroba Charcot-Marie-Tooth
Podľa klasifikácie polyneuropatických syndrómov sa peroneálna (fibulárna) svalová atrofia, Charcot-Marie-Toothova neurónová amyotrofia alebo Charcot-Marie-Toothova choroba (skrátene CMT) vzťahuje na geneticky podmienené motoricko-senzorické polyneuropatie. [ 2 ]
To znamená, že príčinami jeho výskytu sú genetické mutácie. A v závislosti od povahy genetických odchýlok sa rozlišujú hlavné typy alebo druhy tohto syndrómu: demyelinizačný a axonálny. Prvá skupina zahŕňa Charcot-Marie-Toothov syndróm typu 1 (CMT1), ktorý vzniká v dôsledku duplikácie génu PMP22 na chromozóme 17, ktorý kóduje transmembránový periférny myelínový proteín 22. V dôsledku toho dochádza k segmentálnej demyelinizácii axónového obalu (výbežky nervových buniek) a k zníženiu rýchlosti vedenia nervového signálu. Okrem toho môžu byť mutácie aj v niektorých ďalších génoch.
Axonálna forma je Charcot-Marie-Toothova choroba typu 2 (CMT2), ktorá postihuje samotné axóny a je spojená s patologickými zmenami v géne MFN2 v lokuse 1p36.22, kódujúcom membránový proteín mitofusín-2, ktorý je nevyhnutný pre fúziu mitochondrií a tvorbu funkčných mitochondriálnych sietí vo vnútri periférnych nervových buniek. Existuje viac ako tucet podtypov CMT2 (s mutáciami v špecifických génoch).
Treba poznamenať, že v súčasnosti bolo identifikovaných viac ako sto génov, ktorých poškodenie, prenášané dedičnosťou, spôsobuje rôzne podtypy Charcot-Marie-Toothovej choroby. Napríklad mutácie v géne RAB7 vedú k CMT typu 2B; zmena génu SH3TC2 (kódujúceho jeden z membránových proteínov Schwannových buniek) spôsobuje CMT typu 4C, ktorá sa prejavuje v detstve a je charakterizovaná demyelinizáciou motorických a senzorických neurónov (existuje tucet a pol foriem typu 4 tohto ochorenia).
Zriedkavá CMT typu 3 (nazývaná Dejerine-Sottasov syndróm) sa začína vyvíjať v ranom detstve a je spôsobená mutáciami v génoch PMP22, MPZ, EGR2 a ďalších.
Keď sa CMT typu 5 vyskytne vo veku 5-12 rokov, pozoruje sa nielen motorická neuropatia (vo forme spastickej paraparézy dolných končatín), ale aj poškodenie zrakového a sluchového nervu.
Svalová slabosť a optická atrofia (so stratou zraku), ako aj problémy s rovnováhou sú typické pre CMT typu 6. A pri Charcot-Marie-Toothovej chorobe typu 7 sa nielen vyskytuje motoricko-senzorická neuropatia, ale aj ochorenie sietnice vo forme retinitis pigmentosa.
U mužov sa častejšie vyskytuje X-viazaná CMT alebo Charcot-Marie-Toothova choroba s tetraparézou končatín (slabosť pohybu rúk aj nôh), čo je demyelinizačný typ a predpokladá sa, že je výsledkom mutácie v géne GJB1 na dlhom ramene chromozómu X, ktorý kóduje konexín 32, transmembránový proteín Schwannových buniek a oligodendrocytov, ktorý reguluje prenos nervových signálov. [ 3 ]
Rizikové faktory
Hlavným rizikovým faktorom pre CMT je rodinná anamnéza ochorenia, teda u blízkych príbuzných.
Podľa genetikov, ak sú obaja rodičia nositeľmi autozomálne recesívneho génu pre Charcot-Marie-Toothovu chorobu, riziko narodenia dieťaťa, u ktorého sa toto ochorenie vyvinie, je 25 %. A riziko, že dieťa bude nositeľom tohto génu (ale nebude mať žiadne príznaky), sa odhaduje na 50 %.
V prípade dedičnosti viazanej na chromozóm X (keď sa mutovaný gén nachádza na chromozóme X ženy) existuje 50 % riziko, že matka prenesie gén na svojho syna, u ktorého sa vyvinie karcinóm meningitídy (CMT). Ochorenie sa nemusí prejaviť pri narodení dievčatka, ale synovia (vnúčatá) dcéry môžu zdediť chybný gén a ochorenie sa u nich vyvinie.
Patogenézy
Pri akomkoľvek type Charcot-Marie-Toothovej choroby je jej patogenéza spôsobená dedičnou anomáliou periférnych nervov: motorických (pohybových) a senzorických (citlivých).
Ak je typ CMT demyelinizačný, potom deštrukcia alebo defekt myelínového obalu, ktorý chráni axóny periférnych nervov, vedie k spomaleniu prenosu nervových impulzov v periférnom nervovom systéme - medzi mozgom, svalmi a zmyslovými orgánmi.
Pri axonálnom type ochorenia sú postihnuté samotné axóny, čo negatívne ovplyvňuje silu nervových signálov, ktorá je nedostatočná pre plnú stimuláciu svalov a zmyslových orgánov.
Prečítajte si tiež:
Ako sa prenáša Charcot-Marie-Toothov syndróm? Poškodené gény sa môžu dediť autozomálne dominantne alebo autozomálne recesívne.
Najbežnejší typ, autozomálne dominantná dedičnosť, sa vyskytuje, keď existuje jedna kópia mutovaného génu (ktorý nosí jeden z rodičov). Pravdepodobnosť prenosu CMT na každé narodené dieťa sa odhaduje na 50 %. [ 4 ]
Pri autozomálne recesívnej dedičnosti sú na rozvoj ochorenia potrebné dve kópie defektného génu (jeden od každého rodiča, ktorý nevykazuje žiadne príznaky ochorenia).
V 40 – 50 % prípadov sa vyskytuje autozomálne dominantná dedičná demyelinizácia, t. j. karcinómová melanokarcinómová patológia typu 1; v 12 – 26 % prípadov axonálna karcinómová melanokarcinómová patológia, t. j. typ 2. A v 10 – 15 % prípadov sa pozoruje dedičnosť viazaná na chromozóm X. [ 5 ]
Príznaky Choroba Charcot-Marie-Tooth
Prvé príznaky tohto ochorenia sa zvyčajne začínajú objavovať v detstve a dospievaní a postupne sa rozvíjajú počas celého života, hoci syndróm sa môže prejaviť aj neskôr. Kombinácia symptómov je variabilná a rýchlosť progresie ochorenia, ako aj jeho závažnosť, nie je možné predpovedať.
Medzi typické príznaky počiatočného štádia patrí zvýšená celková únava; znížený tonus (slabosť) svalov chodidiel, členkov a holení; nedostatok reflexov. To sťažuje pohyb chodidiel a vedie k dysbázii (porucha chôdze) vo forme vyššieho zdvihnutia nôh, často s častými zakopnutiami a pádmi. Medzi príznaky Charcot-Marie-Toothovej choroby u malého dieťaťa môže patriť výrazná nemotornosť a pre vek netypické ťažkosti s chôdzou spojené s obojstranným poklesom chodidla. Charakteristické sú aj deformity chodidiel: vysoká klenba (dutá noha) alebo výrazné ploché nohy, zakrivené (kladivkovité) prsty.
V prípade chôdze po špičkách na pozadí svalovej hypotónie môže neurológ mať podozrenie, že dieťa má CMT typu 4, pri ktorej deti nemusia byť schopné chodiť do adolescencie.
S postupom ochorenia sa svalová atrofia a slabosť šíria do horných končatín, čo sťažuje vykonávanie jemnej motoriky a bežných úkonov s rukami. Znížené hmatové vnemy a schopnosť cítiť teplo a chlad, ako aj necitlivosť v nohách a rukách, naznačujú poškodenie axónov senzorických nervov.
Pri Charcot-Marie-Toothovej chorobe typu 3 a 6, ktorá sa prejavuje v detstve, sa pozoruje senzorická ataxia (zhoršená koordinácia pohybov a rovnováhy), svalové zášklby a tremor, poškodenie tvárového nervu, atrofia zrakového nervu s nystagmom a strata sluchu.
V neskorších štádiách sa môže vyskytnúť nekontrolovateľné trasenie (tremor) a časté svalové kŕče; problémy s pohybom môžu viesť k rozvoju bolesti: svalovej, kĺbovej, neuropatickej.
Komplikácie a následky
Charcot-Marie-Toothova choroba môže mať komplikácie a následky, ako napríklad:
- častejšie výrony a zlomeniny;
- kontraktúry spojené so skrátením periartikulárnych svalov a šliach;
- skolióza (zakrivenie chrbtice);
- problémy s dýchaním – keď sú poškodené nervové vlákna, ktoré inervujú svaly bránice:
- strata schopnosti samostatného pohybu.
Diagnostika Choroba Charcot-Marie-Tooth
Diagnóza zahŕňa klinické vyšetrenie, anamnézu (vrátane rodinnej anamnézy), neurologické a systémové vyšetrenie.
Vykonávajú sa testy na kontrolu rozsahu pohybu, citlivosti a šľachových reflexov. Nervové vedenie sa dá posúdiť pomocou inštrumentálnej diagnostiky – elektromyografie alebo elektroneuromyografie. Môže byť potrebný aj ultrazvuk alebo magnetická rezonancia. [ 6 ]
Genetické alebo DNA testovanie na detekciu najbežnejších genetických mutácií, ktoré spôsobujú karcinóm meningitídy (CMT) vo vzorke krvi, je obmedzené, pretože DNA testy nie sú v súčasnosti dostupné pre všetky typy ochorenia. Viac informácií nájdete v časti Genetické testovanie.
V niektorých prípadoch sa vykonáva biopsia periférneho nervu (zvyčajne surálneho nervu).
Odlišná diagnóza
Diferenciálna diagnostika zahŕňa iné periférne neuropatie, Duchennovu svalovú dystrofiu, myelopatické a myastenické syndrómy, diabetickú neuropatiu, myelopatie pri skleróze multiplex a amyotrofickej laterálnej skleróze, Guillain-Barréov syndróm, traumu peroneálneho nervu a jeho atrofiu (vrátane zovretia medzi bedrovými platničkami chrbtice), poškodenie mozočka alebo talamu, ako aj vedľajšie účinky chemoterapie (počas liečby cytostatikami, ako je vinkristín alebo paklitaxel). [ 7 ]
Komu sa chcete obrátiť?
Liečba Choroba Charcot-Marie-Tooth
Liečba tohto dedičného ochorenia dnes zahŕňa cvičebnú terapiu (zameranú na posilnenie a natiahnutie svalov); ergoterapiu (ktorá pomáha pacientom so svalovou slabosťou v rukách); a používanie ortopedických pomôcok na uľahčenie chôdze. V prípade potreby sa predpisujú lieky proti bolesti alebo antikonvulzíva. [ 8 ]
V prípadoch ťažkých plochých nôh sa môže vykonať osteotómia a v prípade deformácie päty je indikovaná chirurgická korekcia – artrodéza. [ 9 ]
Prebieha výskum genetickej zložky ochorenia aj metód jeho liečby. Použitie kmeňových buniek, niektorých hormónov, lecitínu alebo kyseliny askorbovej zatiaľ neprinieslo pozitívne výsledky.
Vďaka nedávnemu výskumu sa však v blízkej budúcnosti môže skutočne objaviť niečo nové v liečbe Charcot-Marie-Toothovej choroby. Francúzska spoločnosť Pharnext tak od roku 2014 vyvíja a od polovice roka 2019 prebiehajú klinické skúšky lieku PXT3003 na liečbu CMT typu 1 u dospelých, ktorý potláča zvýšenú expresiu génu PMP22, zlepšuje myelinizáciu periférnych nervov a zmierňuje neuromuskulárne symptómy.
Špecialisti z lekárskej spoločnosti Sarepta Therapeutics (USA) pracujú na vytvorení génovej terapie pre Charcot-Marie-Toothovu chorobu typu 1. Táto terapia bude využívať neškodný adeno-asociovaný vírus (AAV) rodu Dependovirus s lineárnym jednovláknovým DNA genómom, ktorý prenesie do tela gén NTF3 kódujúci proteín neurotrofín-3 (NT-3) potrebný pre fungovanie Schwannových nervových buniek.
Spoločnosť Helixmith začne do konca roka 2020 klinické skúšky génovej terapie Engensis (VM202) vyvinutej v Južnej Kórei na liečbu svalových symptómov pri karpometakarpálnej melanokarcíne typu 1. [ 10 ]
Prevencia
Prevenciou CMT môže byť genetické poradenstvo budúcich rodičov, najmä ak má niekto z páru rodinnú anamnézu tohto ochorenia. Boli však identifikované aj prípady de novo bodových génových mutácií, teda pri absencii ochorenia v rodinnej anamnéze.
Počas tehotenstva je možné pravdepodobnosť Charcot-Marie-Toothovej choroby u budúceho dieťaťa skontrolovať biopsiou choriových klkov (od 10. do 13. týždňa tehotenstva), ako aj analýzou plodovej vody (v 15. až 18. týždni).
Predpoveď
Vo všeobecnosti prognóza rôznych typov Charcot-Marie-Toothovej choroby závisí od klinickej závažnosti, ale vo všetkých prípadoch choroba postupuje pomaly. Mnohí pacienti majú postihnutie, hoci to neznižuje dĺžku života.