
Všetok obsah iLive je lekársky kontrolovaný alebo kontrolovaný, aby sa zabezpečila čo najväčšia presnosť faktov.
Máme prísne smernice týkajúce sa získavania zdrojov a len odkaz na seriózne mediálne stránky, akademické výskumné inštitúcie a vždy, keď je to možné, na lekársky partnerské štúdie. Všimnite si, že čísla v zátvorkách ([1], [2] atď.) Sú odkazmi na kliknutia na tieto štúdie.
Ak máte pocit, že niektorý z našich obsahov je nepresný, neaktuálny alebo inak sporný, vyberte ho a stlačte kláves Ctrl + Enter.
Huntingtonova choroba
Lekársky expert článku

Posledná kontrola: 05.07.2025
Huntingtonova choroba je autozomálne dominantné neurodegeneratívne ochorenie charakterizované progresívnym kognitívnym poklesom, mimovoľnými pohybmi a zhoršenou motorickou koordináciou, ktorá sa začína v strednom veku. Diagnóza sa potvrdzuje genetickým testovaním. Liečba je primárne symptomatická. Genetické testovanie sa môže odporučiť pokrvným príbuzným. George Huntington prvýkrát opísal tento stav v roku 1872 po štúdiu rodinného prípadu u obyvateľov Long Islandu.
Prevalencia Huntingtonovej choroby je približne 10 prípadov na 100 000 obyvateľov a vzhľadom na jej neskorý nástup má približne 30 ľudí zo 100 000 50 % riziko, že sa u nich počas života vyvinie. Hoci sa ochorenie najčastejšie objavuje medzi 35. a 40. rokom života, vekové rozpätie nástupu je pomerne široké, pričom najskorší nástup je vo veku 3 rokov a najneskorší vo veku 90 rokov. Hoci sa pôvodne predpokladalo, že ochorenie má 100 % penetranciu, v súčasnosti sa predpokladá, že to tak nie je vždy. U jedincov, ktorí zdedili gén pre toto ochorenie po otcovi, sa ochorenie prejaví v priemere o 3 roky skôr ako u tých, ktorí zdedili patologický gén po matke. U približne 80 % pacientov, ktorí zdedili patologický gén po otcovi, sa ochorenie prejaví pred dovŕšením 20. roku života. Fenomén skoršieho prejavu genetickej chyby u potomstva sa nazýva anticipácia.
 [ 1 ]
[ 1 ]
Čo spôsobuje Huntingtonovu chorobu?
Huntingtonova choroba nemá rodovú preferenciu. Prejavuje sa atrofia nucleus caudatus, kde malé neuróny degenerujú a hladina neurotransmiterov - kyseliny gama-aminomaslovej (GABA) a substancie P - klesá.
Za rozvoj Huntingtonovej choroby je zodpovedný mutantný gén so zvýšeným počtom („expanziou“) sekvencií DNA CAG (cysteín-alanín-glycín) kódujúcich aminokyselinu glutamín. Produkt tohto génu, veľký proteín huntingtín, obsahuje nadmerné množstvo polyglutamínových zvyškov, čo vedie k ochoreniu neznámym mechanizmom. Čím viac opakovaní CAG, tým skôr ochorenie debutuje a tým je jeho priebeh závažnejší. Z generácie na generáciu sa môže počet opakovaní zvyšovať, čo časom vedie k zhoršeniu rodinného fenotypu.
Napriek značnému záujmu o genetické a biochemické zmeny pri Parkinsonovej chorobe bolo hľadanie génu pre túto chorobu neúspešné až do konca 70. rokov 20. storočia. V tom čase Nancy Wexlerová a Allan Tobin zorganizovali workshop sponzorovaný Nadáciou pre dedičné choroby, aby prediskutovali stratégiu hľadania génu pre Huntingtonovu chorobu. David Housman, David Botstein a Ray White, ktorí sa stretnutia zúčastnili, navrhli, že nedávno vyvinuté techniky rekombinantnej DNA by mohli pomôcť dosiahnuť tento cieľ. Kľúčovou úlohou projektu bolo nájsť veľkú rodinu s mnohými generáciami Huntingtonovej choroby, aby sa získali vzorky DNA. V roku 1979 sa začal spoločný projekt vedcov z Venezuely a Spojených štátov zameraný na vyšetrenie veľkej rodiny s Huntingtonovou chorobou žijúcej na brehu jazera Maracheibo (Venezuela). V roku 1983 bol gén Huntingtonovej choroby lokalizovaný na konci krátkeho ramena chromozómu 4 (Gusella a kol., 1983) a o desaťročie neskôr sa zistilo, že mutácia tohto génu spočíva vo zvýšení počtu opakovaní trinukleotidu cytozín-adenín-guanín (CAG) (Huntington's Disease Collaborative Research Group, 1993). Metodika vyvinutá touto vedeckou skupinou sa v súčasnosti považuje za štandard pre pozičné klonovanie nových génov.
Zatiaľ čo gén divokého typu má úsek 10 – 28 CAG opakovaní, mutantná forma génu, ktorý spôsobuje Huntingtonovu chorobu, má zvýšený úsek z 39 na viac ako 100 CAG opakovaní. Objav expanzie trinukleotidových opakovaní pomohol vysvetliť mnohé klinické znaky ochorenia. Najmä sa zistila inverzná korelácia medzi vekom nástupu ochorenia a dĺžkou oblasti s opakovanými trinukleotidmi. Predpokladanie otcovskej dedičnosti možno vysvetliť skutočnosťou, že u mužov sa počas spermatogenézy často vyskytuje zvýšenie počtu opakovaní. Analýza nových mutácií ukázala, že sa zvyčajne vyskytujú, keď jeden z rodičov, zvyčajne otec, mal počet CAG opakovaní vyšší ako 28; v tomto prípade sa počet týchto opakovaní v ďalšej generácii zvýšil. Teraz sa zistilo, že ak počet opakovaní nie je vyšší ako 28, stabilne sa prenáša z generácie na generáciu. Ak je počet opakovaní od 29 do 35, potom sa príznaky Huntingtonovej choroby neprejavujú, ale pri prenose na potomstvo sa dĺžka tejto oblasti môže zväčšiť. Ak je počet opakovaní od 36 do 39, potom sa v niektorých prípadoch (ale nie vždy) ochorenie môže prejaviť klinicky (neúplná penetrancia) a pri prenose na potomstvo je možné zvýšenie počtu trinukleotidových opakovaní. Ak počet opakovaní presiahne 40, potom sa ochorenie vyskytuje takmer vo všetkých prípadoch a pri prenose na potomstvo je možné ďalšie rozšírenie opakovaní. Dôvody zvýšenia počtu opakovaní zostávajú neznáme.
Patomorfológia Huntingtonovej choroby
Huntingtonova choroba sa vyznačuje stratou neurónov prevažne v nucleus caudatus a putamene a do istej miery aj v kortexe a iných mozgových štruktúrach. Celková hmotnosť mozgu pri Huntingtonovej chorobe sa znižuje nielen znížením počtu neurónov, ale aj stratou bielej hmoty. V mozgovej kôre sú najviac postihnuté bunky vo vrstvách V a VI. Závažnosť mikro- a makroskopických degeneratívnych zmien (upravených podľa veku v čase úmrtia) koreluje s počtom CAG opakovaní. Podrobná patologická analýza zmien v niekoľkých stovkách prípadov Huntingtonovej choroby ukázala, že degenerácia striata začína v dorzomediálnej časti nucleus caudatus a dorsolaterálnej časti putamenu a potom sa šíri ventrálne. Rôzne skupiny neurónov v nucleus caudatus a putamene sú postihnuté v rôznej miere. Interneuróny v striate zostávajú relatívne neporušené, ale niektoré projekčné neuróny sú selektívne postihnuté. Pri juvenilnej forme Huntingtonovej choroby sú patomorfologické zmeny v striate výraznejšie a rozšírenejšie, postihujú mozgovú kôru, mozoček, talamus a globus pallidus.
Neurochemické zmeny pri Huntingtonovej chorobe
GABA. Neurochemické štúdie mozgu u pacientov s Huntingtonovou chorobou odhalili významný pokles koncentrácie GABA v striate. Následné štúdie potvrdili, že Huntingtonova choroba je spojená so znížením počtu GABAergných neurónov a ukázali, že koncentrácie GABA sú znížené nielen v striate, ale aj v jeho projekčných zónach - vonkajších a vnútorných segmentoch globus pallidus a substantia nigra. V mozgu pri Huntingtonovej chorobe boli pomocou štúdií väzby receptorov a in situ hybridizácie mRNA detegované aj zmeny v GABA receptoroch. Počet GABA receptorov bol mierne znížený v nucleus caudatus a putamene, ale zvýšený v retikulárnej časti substantia nigra a vonkajšom segmente globus pallidus, čo je pravdepodobne spôsobené denervačnou precitlivenosťou.
Acetylcholín. Acetylcholín sa používa ako neurotransmiter veľkými neostnatými interneurónmi v striate. Včasné postmortálne štúdie u pacientov s Huntingtonovou chorobou preukázali zníženú aktivitu cholín acetyltransferázy (ChAT) v striate, čo naznačuje stratu cholinergných neurónov. Avšak v porovnaní s významným znížením GABAergných neurónov sú cholinergné interneuróny relatívne ušetrené. Preto je hustota neurónov pozitívnych na acetylcholínesterázu a aktivita ChAT v striate v skutočnosti relatívne zvýšená v porovnaní s kontrolami rovnakého veku.
Látka P. Látka P je obsiahnutá v mnohých stredne ostnatých neurónoch striata, ktoré prevažne vyčnievajú do vnútorného segmentu globus pallidus a substantia nigra a zvyčajne obsahujú aj dynorfín a GABA. Hladiny látky P v striate a pars reticularis substantia nigra sú pri Huntingtonovej chorobe znížené. V terminálnom štádiu ochorenia imunohistochemické štúdie odhalili významné zníženie počtu neurónov obsahujúcich látku P. V skorších štádiách sú neuróny obsahujúce látku P a vyčnievajúce do vnútorného segmentu globus pallidus relatívne ušetrené v porovnaní s neurónmi vyčnievajúcimi do pars reticularis substantia nigra.
Opioidné peptidy. Enkefalín je obsiahnutý v stredne ostnatých projekčných GABAergných neurónoch nepriamej dráhy, ktoré vyčnievajú do vonkajšieho segmentu globus pallidus a nesú D2 receptory. Imunohistochemické štúdie ukázali, že neuróny obsahujúce enkefalín vyčnievajúce do vonkajšieho segmentu globus pallidus sa pri Huntingtonovej chorobe strácajú už na začiatku liečby. Tieto bunky zrejme odumierajú skôr ako bunky obsahujúce substanciu P vyčnievajúce do vnútorného segmentu globus pallidus.
Katecholamíny. Neuróny obsahujúce biogénne amíny (dopamín, serotonín) a vyčnievajúce do striata sa nachádzajú v kompaktnej časti substantia nigra, ventrálnom tegmente a jadrách raphe. Zatiaľ čo noradrenergné projekcie do ľudského striata sú minimálne, hladiny serotonínu a dopamínu (na gram tkaniva) v striate sú zvýšené, čo naznačuje zachovanie týchto aferentných projekcií napriek výraznej strate vlastných neurónov striata. Dopaminergné neuróny substantia nigra zostávajú neporušené pri klasických aj juvenilných formách Huntingtonovej choroby.
Somatostatín/neuropeptid Y a syntetáza oxidu dusnatého. Meranie hladín somatostatínu a neuropeptidu Y v striate pri Huntingtonovej chorobe odhalilo 4-5-násobné zvýšenie v porovnaní s normálnymi tkanivami. Imunohistochemické štúdie preukázali absolútnu zachovanie striatálnych interneurónov obsahujúcich neuropeptid Y, somatostatín a syntetázu oxidu dusnatého. Tieto neuróny sú teda rezistentné voči patologickému procesu.
Excitačné aminokyseliny. Predpokladá sa, že selektívna bunková smrť pri Huntingtonovej chorobe je spôsobená neurotoxickým účinkom vyvolaným glutamátom. Hladiny glutamátu a kyseliny chinolínovej (endogénny neurotoxín, ktorý je vedľajším produktom metabolizmu serotonínu a agonistom glutamátových receptorov) v striate pri Huntingtonovej chorobe sú mierne zmenené, ale nedávna štúdia s použitím MR spektroskopie odhalila zvýšenie hladín glutamátu in vivo. Hladina gliového enzýmu zodpovedného za syntézu kyseliny chinolínovej v striate pri Huntingtonovej chorobe je približne 5-krát zvýšená v porovnaní s normálom, zatiaľ čo aktivita enzýmu, ktorý zabezpečuje degradáciu kyseliny chinolínovej, je pri Huntingtonovej chorobe zvýšená len o 20 – 50 %. Syntéza kyseliny chinolínovej teda môže byť pri Huntingtonovej chorobe zvýšená.
Štúdie receptorov excitačných aminokyselín (EAA) pri Huntingtonovej chorobe odhalili významné zníženie počtu NMDA, AMPA, kainátových a metabotropných glutamátových receptorov v striate, ako aj AMPA a kainátových receptorov v mozgovej kôre. V neskorom štádiu Huntingtonovej choroby boli NMDA receptory prakticky neprítomné, zatiaľ čo v predklinických a skorých štádiách sa zaznamenalo významné zníženie počtu týchto receptorov.
Selektívna citlivosť. Pri Huntingtonovej chorobe sa selektívne strácajú určité typy striatálnych buniek. Stredne ostnaté neuróny, ktoré vyčnievajú do vonkajšieho segmentu globus pallidus a obsahujú GABA a enkefalín, odumierajú veľmi skoro v priebehu ochorenia, rovnako ako neuróny obsahujúce GABA a substanciu P a vyčnievajúce do retikulárnej časti substantia nigra. Strata neurónov obsahujúcich GABA a enkefalín a vyčnievajúcich do vonkajšieho segmentu globus pallidus deinhibuje túto štruktúru, čo následne vedie k aktívnej inhibícii subtalamického jadra. Znížená aktivita subtalamického jadra môže zrejme vysvetliť choreiformné pohyby, ku ktorým dochádza pri Huntingtonovej chorobe. Dlho je známe, že fokálne lézie subtalamického jadra môžu spôsobiť choreu. Strata neurónov GABA a substancie P vyčnievajúcich do pars reticularis substantia nigra je pravdepodobne zodpovedná za okulomotorické poruchy pozorované pri Huntingtonovej chorobe. Táto dráha normálne inhibuje neuróny pars reticularis z substantia nigra, ktoré vyčnievajú do superior colliculus, a ktoré následne regulujú sakády. Pri juvenilnej Huntingtonovej chorobe sú vyššie uvedené dráhy postihnuté závažnejšie a navyše sa striatálne projekcie do vnútorného segmentu globus pallidus strácajú skoro.
Proteín huntingtín, kódovaný génom, ktorého mutácia spôsobuje Huntingtonovu chorobu, sa nachádza v rôznych štruktúrach mozgu a iných tkanivách. Huntingtín sa bežne nachádza prevažne v cytoplazme neurónov. Proteín sa nachádza vo väčšine neurónov v mozgu, ale nedávne údaje ukazujú, že jeho obsah je vyšší v matrixových neurónoch ako v striozomálnych neurónoch a vyšší v projekčných neurónoch ako v interneurónoch. Selektívna citlivosť neurónov teda koreluje s ich obsahom huntingtínu, ktorý je bežne prítomný v určitých neuronálnych populáciách.
Rovnako ako v mozgoch pacientov s Huntingtonovou chorobou, aj u myší transgénnych pre N-terminálny fragment génu Huntingtonovej choroby so zvýšeným počtom opakovaní, huntingtín tvorí husté agregáty v jadrách neurónov. Tieto intranukleárne inklúzie sa tvoria v neurónoch striatálnej projekcie (ale nie v interneurónoch). U transgénnych myší sa inklúzie tvoria niekoľko týždňov pred nástupom príznakov. Tieto údaje naznačujú, že huntingtínový proteín obsahujúci zvýšený počet glutamínových zvyškov, ktorých inklúzie kódujú trinukleotidové opakovania, alebo jeho fragment, sa hromadí v jadre a následne môže narušiť jeho kontrolu bunkových funkcií.
Príznaky Huntingtonovej choroby
Vek, v ktorom sa u pacientov s Huntingtonovou chorobou objavili prvé príznaky, je ťažké presne určiť, pretože ochorenie sa prejavuje postupne. Zmeny osobnosti a správania, mierne poruchy koordinácie sa môžu vyskytnúť mnoho rokov pred objavením sa zjavnejších príznakov. V čase stanovenia diagnózy má väčšina pacientov choreické pohyby, zhoršenú koordináciu jemných pohybov a pomalé generovanie vôľových sakád. S progresiou ochorenia sa zhoršuje schopnosť organizovať si činnosti, znižuje sa pamäť, ťažkosti s rečou, zhoršujú sa okulomotorické poruchy a zhoršený výkon koordinovaných pohybov. Hoci v skorom štádiu ochorenia nedochádza k žiadnym zmenám svalov a držania tela, s postupom ochorenia sa môžu vyvinúť dystonické držanie tela, ktoré sa časom môžu zmeniť na dominantný príznak. V neskorom štádiu sa reč stáva nezrozumiteľnou, prehĺtanie sa stáva výrazne ťažkým, chôdza sa stáva nemožnou. Huntingtonova choroba zvyčajne postupuje 15 – 20 rokov. V terminálnom štádiu je pacient bezmocný a vyžaduje si neustálu starostlivosť. Smrteľný výsledok nie je priamo spojený s primárnym ochorením, ale s jeho komplikáciami, napríklad so zápalom pľúc.
Demencia pri Huntingtonovej chorobe
Kód MKCH-10
P02.2. Demencia pri Huntingtonovej chorobe (G10).
Demencia sa vyvíja ako jeden z prejavov systémového degeneratívne-atrofického procesu s prevažným poškodením striatálneho systému mozgu a ďalších subcekálnych jadier. Dedí sa autozomálne dominantným spôsobom.
Choroba sa spravidla prejavuje v tretej alebo štvrtej dekáde života choreoformnou hyperkinézou (najmä v oblasti tváre, rúk, ramien, chôdze), zmenami osobnosti (excitabilné, hysterické a schizoidné typy osobnostných anomálií), psychotickými poruchami (špeciálna depresia s pochmúrnosťou, mrzutosťou, dysfóriou; paranoidná nálada).
Pre diagnostiku má osobitný význam kombinácia choreoformnej hyperkinézy, demencie a dedičnej záťaže. Pre túto demenciu je špecifické nasledovné:
- pomalý postup (v priemere 10 – 15 rokov): disociácia medzi zostávajúcou schopnosťou postarať sa o seba a zjavnou intelektuálnou neschopnosťou v situáciách vyžadujúcich produktívnu duševnú prácu (koncepčné myslenie, učenie sa nových vecí);
- výrazná nerovnomernosť duševnej výkonnosti, ktorá je založená na hrubých poruchách pozornosti a nestálosti postojov pacienta („trhavé“ myslenie, podobné hyperkinéze);
- atypickosť zjavných porušení vyšších kortikálnych funkcií;
- inverzný vzťah medzi nárastom demencie a závažnosťou psychotických porúch.
Vzhľadom na vysoký podiel psychotických (paranoidné bludy žiarlivosti, prenasledovania) a dysforických porúch v klinickom obraze ochorenia sa liečba vykonáva pomocou rôznych neuroleptík, ktoré blokujú dopaminergné receptory (deriváty fenotiazínu a butyrofenónu) alebo znižujú hladinu dopamínu v tkanivách (rezerpín).
Používa sa haloperidol (2-20 mg/deň), tiaprid (100-600 mg/deň) maximálne tri mesiace, tioridazín (do 100 mg/deň), rezerpín (0,25-2 mg/deň) a antikonvulzívum klonazepam (1,5-6 mg/deň). Tieto lieky pomáhajú znižovať hyperkinézu, vyhladzovať afektívne napätie a kompenzovať poruchy osobnosti.
Ústavná liečba duševných porúch sa vykonáva s ohľadom na vedúci syndróm, vek a celkový stav pacienta. Pri ambulantnej liečbe sú princípy terapie rovnaké (kontinuálna udržiavacia liečba porúch pohybu, periodická zmena lieku). Pri ambulantnej liečbe sa používajú nižšie dávky neuroleptík.
Rehabilitačné opatrenia pre miernu a stredne ťažkú demenciu zahŕňajú ergoterapiu, psychoterapiu a kognitívny tréning. Je potrebné spolupracovať s rodinnými príslušníkmi a poskytovať psychologickú podporu ľuďom starajúcim sa o pacienta. Hlavnou metódou prevencie ochorenia je lekárske a genetické poradenstvo najbližších príbuzných pacienta s odporučením na analýzu DNA pri rozhodovaní o tehotenstve.
Prognóza je vo všeobecnosti nepriaznivá. Priebeh ochorenia je pomaly progresívny a ochorenie zvyčajne vedie k úmrtiu po 10-15 rokoch.
[ 18 ]
Čo vás trápi?
Diagnóza Huntingtonovej choroby
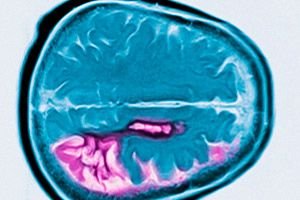
Diagnóza je založená na typických príznakoch, rodinnej anamnéze a genetickom testovaní. V dôsledku atrofie hlavy nucleus caudatus (jadro caudatus) sa pomocou MRI a CG v neskorom štádiu ochorenia odhalí zväčšenie mozgových komôr.
Liečba Huntingtonovej choroby
Liečba Huntingtonovej choroby je symptomatická. Choreu a agitáciu možno čiastočne potlačiť neuroleptikami (napr. chlórpromazín 25 – 300 mg perorálne 3-krát denne, haloperidol 5 – 45 mg perorálne 2-krát denne) alebo rezerpín 0,1 mg perorálne jedenkrát denne. Dávky sa zvyšujú na maximum tolerovanej dávky (predtým, ako sa objavia vedľajšie účinky, ako je ospalosť, parkinsonizmus; pri rezerpíne hypotenzia). Cieľom empirickej terapie je znížiť glutamatergický prenos cez N-metyl-O-aspartátové receptory a udržať produkciu energie v mitochondriách. Liečba zameraná na zvýšenie GABA v mozgu je neúčinná.
Genetické testovanie a poradenstvo sú dôležité, pretože príznaky ochorenia sa objavujú po dosiahnutí plodného veku. Ľudia s pozitívnou rodinnou anamnézou a tí, ktorí majú záujem o testovanie, sú odosielaní do špecializovaných centier, berúc do úvahy všetky etické a psychologické dôsledky.
Symptomatická liečba Huntingtonovej choroby
Neexistuje účinná liečba, ktorá by dokázala zastaviť progresiu Huntingtonovej choroby. Bolo vykonaných niekoľko štúdií s rôznymi liekmi, ale nedosiahol sa žiadny významný účinok. Neuroleptiká a iné antagonisty dopamínových receptorov sa široko používajú na korekciu duševných porúch a mimovoľných pohybov u pacientov s Huntingtonovou chorobou. Mimovoľné pohyby odrážajú nerovnováhu medzi dopaminergným a GABAergným systémom. Preto sa neuroleptiká používajú na zníženie nadmernej dopaminergnej aktivity. Tieto lieky však samotné môžu spôsobiť významné kognitívne a extrapyramídové vedľajšie účinky. Okrem toho, s výnimkou prípadov, keď sa u pacienta vyvinie psychóza alebo agitácia, ich účinnosť nebola preukázaná. Neuroleptiká často spôsobujú alebo zhoršujú dysfágiu alebo iné poruchy pohybu. Neuroleptiká novšej generácie, ako je risperidón, klozapín a olanzapín, môžu byť obzvlášť užitočné pri liečbe Huntingtonovej choroby, pretože spôsobujú menej extrapyramídových vedľajších účinkov, ale môžu zmierniť paranoidné príznaky alebo zvýšenú podráždenosť.
Tetrabenazín a rezerpín tiež znižujú aktivitu dopaminergného systému a môžu zmierniť závažnosť mimovoľných pohybov v skorých štádiách ochorenia. Tieto lieky však môžu spôsobiť depresiu. Keďže samotné ochorenie často spôsobuje depresiu, tento vedľajší účinok výrazne obmedzuje použitie rezerpínu a tetrabenazínu. V neskorších štádiách ochorenia bunky nesúce dopamínové receptory odumierajú, takže účinnosť antagonistov dopamínových receptorov je oslabená alebo stratená.
Neuroleptiká, antidepresíva a anxiolytiká sa používajú na liečbu psychózy, depresie a podráždenosti u pacientov s Huntingtonovou chorobou, ale mali by sa predpisovať len dovtedy, kým pacient tieto príznaky skutočne pretrváva. Lieky, ktoré môžu byť užitočné v jednom štádiu ochorenia, sa môžu stať neúčinnými alebo dokonca škodlivými s postupom ochorenia.
Agonisty GABA receptorov boli testované u pacientov s Huntingtonovou chorobou, pretože sa preukázalo, že Huntingtonova choroba má významný pokles hladín GABA v striate, ako aj precitlivenosť GABA receptorov v jeho projekčných oblastiach. Benzodiazepíny sa ukázali ako účinné v prípadoch, keď sú mimovoľné pohyby a kognitívne poruchy zhoršené stresom a úzkosťou. Mali by sa predpisovať nízke dávky týchto liekov, aby sa predišlo nežiaducim sedatívnym účinkom. U väčšiny pacientov s Huntingtonovou chorobou žiadny z liekov nevedie k významnému zlepšeniu kvality života.
Pri Huntingtonovej chorobe s včasným nástupom a parkinsonovskými príznakmi možno vyskúšať dopaminergné látky, ale ich účinnosť je obmedzená. Okrem toho môže levodopa u týchto pacientov spôsobiť alebo zvýšiť myoklonus. Zároveň môže baklofén u niektorých pacientov s Huntingtonovou chorobou znížiť rigiditu.
[ 26 ], [ 27 ], [ 28 ], [ 29 ]
Preventívna (neuroprotektívna) liečba Huntingtonovej choroby
Hoci je genetický defekt pri Huntingtonovej chorobe známy, zostáva nejasné, ako vedie k selektívnej neuronálnej degenerácii. Predpokladá sa, že preventívne terapie zamerané na zníženie oxidačného stresu a excitotoxicity môžu potenciálne spomaliť alebo zastaviť progresiu ochorenia. Situácia môže byť do istej miery podobná hepatolentikulárnej degenerácii, pri ktorej genetický defekt zostal mnoho rokov neznámy, ale preventívne terapie zamerané na sekundárny účinok, akumuláciu medi, boli „vyliečené“. V tomto ohľade vzbudila osobitnú pozornosť hypotéza, že Huntingtonova choroba je spojená s poruchou energetického metabolizmu a bunkovou smrťou v dôsledku excitotoxicity. Samotné ochorenie môže spôsobiť bunkovú smrť v dôsledku intranukleárnej agregácie N-terminálnych fragmentov huntingtínu, čo narúša bunkové a metabolické funkcie. Tento proces môže postihnúť niektoré skupiny neurónov vo väčšej miere ako iné kvôli ich vyššej citlivosti na excitotoxické poškodenie. V tomto prípade bude preventívna terapia antagonistami receptorov excitačných aminokyselín alebo látkami, ktoré zabraňujú poškodeniu voľnými radikálmi, schopná zabrániť alebo oddialiť nástup a progresiu ochorenia. V laboratórnych modeloch amyotrofickej laterálnej sklerózy sa preukázalo, že antioxidačné látky a antagonisty receptorov (RAA) sú schopné spomaliť progresiu ochorenia. Podobné prístupy môžu byť účinné pri Huntingtonovej chorobe. V súčasnosti prebiehajú klinické skúšky antagonistov glutamátových receptorov a látok, ktoré zvyšujú funkciu komplexu II mitochondriálneho reťazca transportu elektrónov.
[ 30 ], [ 31 ], [ 32 ], [ 33 ], [ 34 ], [ 35 ], [ 36 ], [ 37 ], [ 38 ], [ 39 ]